 К числу самых распространенных наследственных заболеваний кожи связанных с хромосомной нестабильностью, относятся пигментная ксеродерма, синдром Горлина-Гольтца, KID-синдром, врожденный дискератоз, синдром Хабера, синдром Шпиглера-Брока, анемия Фанкони, синдром Ротмунда-Томсона, кожноглазной альбинизм, плоскоклеточная эпителиома Фергюссона-Смита и пигментная ксеродерма.
К числу самых распространенных наследственных заболеваний кожи связанных с хромосомной нестабильностью, относятся пигментная ксеродерма, синдром Горлина-Гольтца, KID-синдром, врожденный дискератоз, синдром Хабера, синдром Шпиглера-Брока, анемия Фанкони, синдром Ротмунда-Томсона, кожноглазной альбинизм, плоскоклеточная эпителиома Фергюссона-Смита и пигментная ксеродерма.
В группу заболеваний, связанных с генетической нестабильностью, включен ряд синдромов, характеризующихся хромосомными повреждениями клеток в ответ на мутагенное действие ультрафиолетового излучения, ионизирующей радиации, химических канцерогенов и онкогенных вирусов. Высокий риск развития злокачественных новообразований при этих заболеваниях обусловлен характерными для них нарушениями репликации нуклеиновых кислот или дефектами репарации.
Ряд наследственных кожных синдромов ассоциируется с опухолями желудочно-кишечного тракта. При этом злокачественные новообразования, как правило, развиваются вследствие малигнизации полипов. Наиболее известными заболеваниями кожи, связанными с опухолями ЖКТ, являются: синдром Гарднера, синдром Пейтца-Егерса, болезнь Каудена синдром Мюир-Торре, синдром Хоуэлла-Эванса, множественная эндокринная неоплазия III типа.
Кожные синдромы, связанные опухолями ЖКТ
Синдром Гарднера — OMIM 175100. Наследственный симпитомокомплекс, включающий различные кожные и костные проявления в сочетании с предраковым интестинальным полипозом толстого кишечника. Тип наследования — аутосомно-доминантный с различной степенью экспрессивности гена, локализованного в хромосоме 5. Первый постоянный признак синдрома, проявляющийся в возрасте от 4 до 10 лет (редко позднее) — эпидермоидные и сально-железистые кисты, десмоидные опухоли, фибромы, липомы, трихоэпителиомы, кератоакантомы, лейомиомы, особенно на коже лица, реже на волосистой части головы, конечностей, груди; остеомы развиваются главным образом в челюстных и клиновидных костях (в 50% случаев); размеры их небольшие, опухоли чаще множественные. Множественные аденоматозные полипы толстого кишечника или только прямой кишки развиваются на 3-4-м десятилетии жизни и могут оставаться бессимптомными до тех пор, пока через много лет не произойдёт их озлокачествление. Гистологически фокусы злокачественной трансформации выявляются в 100% полипов, однако клинически её можно заподозрить у 50-100% больных. Почти в 50% случаев отмечается полипоз желудка и тонкого кишечника, в частности 12-перстной кишки. Иногда наблюдаются фибросаркома, лейомиома желудка или кишечника, а также описаны опухоли щитовидной железы, яичников, надпочечников, печени, меланома. Диагностическую значимость также имеет врождённая гипертрофическая пигментация сетчатки.
Проявления на коже при этом синдроме обычно развиваются задолго до полипоза толстого кишечника, тем самым облегчая его распознавание.
Диагноз основывается на клинических данных и результатах специальных методов исследования пищеварительного тракта — повторной колоноскопии.
Дифференциальный диагноз этого синдрома проводится с кожным заболеванием Каудена, синдромами Пейтца-Егерса, Кронкайна-Канады, Мюир-Торре.
Лечение этого синдрома, связанного опухолями ЖКТ, заключается в раннем профилактическом удалении полипоза толстого кишечника.
Синдром Пейтца-Егерса (син.: периорифициальный лентигиноз) — OMIM 175200. Заболевание, проявляющееся пигментными пятнами, сопровождающимися гамартомами желудочно-кишечного, дыхательного и мочеполового трактов. Мужчины и женщины болеют одинаково часто. Тип наследования этого синдрома поражения кожи аутосомно-доминантный. Локус гена неизвестен. Вероятно, заболевание обусловлено мутацией одного плейотропного гена. Может проявляться в детстве, но чаще всего возникает в юности или ранней молодости. 95% больных имеют характерные пигментные пятна (лентиго, веснушки) тёмно-коричневого цвета круглой или овальной формы, диаметром от 2 до 5 мм на губах (особенно на нижней) или слизистой оболочке щёк, а также вокруг рта, на переносице, в области заднего прохода, реже на кистях и стопах, ладонях, подошвах, в подколенных ямках. Очаги могут быть врождёнными, появляться в младенчестве или детстве и со временем бледнеют, хотя пигментация на слизистой оболочке сохраняется. Также описаны пигментные папилломы слизистой оболочки рта. Иногда наблюдаются гиперпигментация и дистрофия, а также выпадение волос.
Гамартомы (чаще аденоматозные), которые отмечаются у подавляющего большинства больных, значительно чаще поражают тонкий кишечник, реже любой другой орган пищеварительной, дыхательной, мочеполовой системы. Обычно они небольшого размера, округлые, с гладкой поверхностью, сопровождаются приступами боли в животе и желудочно-кишечными кровотечениями, а также склонны к малигнизации. Хотя чаще злокачественные опухоли при этом синдроме отмечаются вне пищеварительного тракта (2-12%) — речь идет о раке яичников, яичек, лёгких, молочной железы. Важная особенность синдрома Пейтца-Егерса — развитие злокачественных новообразований в раннем возрасте (44-48% случаев).
Гистологически в базальном слое эпидермиса обнаруживают увеличение количества меланоцитов, в дерме — скопление меланофоров. Полипы имеют строение доброкачественной аденомы и в 20-25% случаев подвергаются озлокачествлению.
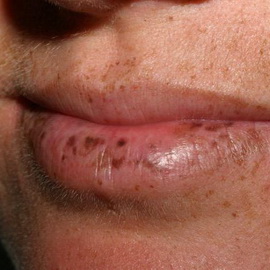 Диагноз устанавливается на основании клиники и результатов гистологического исследования. В целях раннего выявления полипоза проводят рентгенологическое и эндоскопическое исследование.
Диагноз устанавливается на основании клиники и результатов гистологического исследования. В целях раннего выявления полипоза проводят рентгенологическое и эндоскопическое исследование.
Дифференциальный диагноз проводится с веснушками, старческим лентиго, синдромом LEOPARD, наследственными формами лентигиноза, особенно системного, а также с мастоцитозом. Полипы удаляют хирургически.
Течение нередко сопровождается летальным исходом, обусловленным несвоевременно распознанными злокачественными новообразованиями внутренних органов.
Лечение. Пигментацию губ лечат лазерным излучением. Полипы диаметром более 1,5 см, а также кровоточащие полипы удаляют. Каждые 1-3 года больной должен осматриваться гастроэнтерологом и хирургом. Иногда показана профилактическая колонэктомия.
Болезнь Каудена — MIM 158350. Редкий наследственный синдром с аутосомно-доминантным путём передачи (локус гена неизвестен), получивший своё название по фамилии семьи, в которой впервые были выявлены такие больные. Ген картирован на хромосоме 10q23.31, найдены мутации в гене PTEN, которые встречаются у 80% пациентов с болезнью Каудена. Проявляется в возрасте от 4 до 75 лет, в половине случаев до 40 лет. Мужчины болеют чаще.
Характеризуется множественными гамартомами нейроэктодермального, мезодермального и эктодермального происхождения в ассоциации со злокачественными опухолями молочной (нередко двусторонние), щитовидной желёз, желудочно-кишечного тракта (пищевод, двенадцатиперстная и ободочная кишка и т.д.), предстательной железы, матки. При болезни Каудена также обнаруживаются: кистозно-фиброзная мастопатия с ранней злокачественной трансформацией (у мужчин — гинекомастия), зоб, полипоз желудочно-кишечного тракта, поражение центральной нервной системы (умственная отсталость, эпилепсия, невриномы, ганглионевромы, менингиомы височной кости), нарушения менструального цикла, нарушения опорно-двигательной системы (увеличение размера черепа, кифосколиоз, готическое нёбо).
Кожные опухоли представлены преимущественно (89%) трихилеммомами -мелкими папулами, похожими на плоские бородавки, а также другими доброкачественными опухолями придатков кожи, цвет которых может не отличаться от окружающей здоровой кожи, быть розовым или коричневым; они локализуются в центральной части лица, вокруг рта, на губах, ушных раковинах. Иногда множественность трихилеммом уродует больного. Они обычно предшествуют развитию рака внутренних органов. Кроме того, при болезни Каудена могут возникать папилломы дёсен и акральный кератоз (напоминающий акрокератоз Гопфа), базалиомы, плоскоклеточный рак кожи, меркелиома, меланома; липома, ангиома, папилломатоз губ, веррукозная гиперплазия дёсен.
Течение сопровождается возникновением новых опухолей на протяжении всей жизни.
Диагноз устанавливается на основании анамнеза, клинической картины, данных лучевой диагностики (маммография, ультразвуковое исследование щитовидной железы), выявления трихолеммом при гистологическом исследовании. Важна ранняя диагностика трихолеммом как возможного маркёра повышенного риска развития рака молочной железы (20%).
Дифференциальный диагноз проводится с множественными сирингомами, множественными трихоэпителиомами, плоскими бородавками, синдромом Мюир-Торре, гиалинозом кожи и слизистых оболочек.
При лечении опухолей кожи используют лучи лазера или электрокоагуляцию. Необходимо медико-генетическое консультирование, наблюдение у онколога с целью исключения рака молочной и щитовидной железы. Высокий риск развития рака молочной железы иногда требует проведения профилактической двусторонней мастэктомии.
 Мюир-Торре синдром — MIM 158320. Редкое заболевание с аутосомно-доминантным типом наследования, характеризующееся сочетанием опухолей кожи и множественных новообразований внутренних органов. Проявляется, на 5-6-м десятилетиях жизни. Ген локализован в хромосомах 3р21.3 и 2р22-р211. Генетическая основа заболевания не ясна. Предполагаете: что синдром является вариантом семейного ракового синдрома Линча (с мутациями в гене MSH1). Потомки родственников с синдромом Линча страдали синдромом Мюир-Торре и имели мутации в гене MSH2.
Мюир-Торре синдром — MIM 158320. Редкое заболевание с аутосомно-доминантным типом наследования, характеризующееся сочетанием опухолей кожи и множественных новообразований внутренних органов. Проявляется, на 5-6-м десятилетиях жизни. Ген локализован в хромосомах 3р21.3 и 2р22-р211. Генетическая основа заболевания не ясна. Предполагаете: что синдром является вариантом семейного ракового синдрома Линча (с мутациями в гене MSH1). Потомки родственников с синдромом Линча страдали синдромом Мюир-Торре и имели мутации в гене MSH2.
Среди кожных опухолей, которые чаще располагаются на лице, помимо множественных аденом и рака сальных желёз встречаются: кератоакантомы, АК, базалиомы. Примерно в половине случаев они возникают после развития злокачественных новообразований внутренних органов (аденокарцинома толстой кишки, рак желудка, тощей, двенадцатиперстной кишки, гортани мочевого пузыря, почки, предстательной железы, эндометрия, гортани, вульвы, яичников, лёгких). Новообразования внутренних органов при синдроме Мюир-Торре отличаются низкой степенью злокачественности, поэтому продолжительность жизни больных с этим заболеванием выше, чем у больных с аналогичной локализацией рака.
Диагноз этого кожного синдрома основывается на сочетании кожных новообразований и опухолей внутренних органов. Примерно в 30% случаев кожные опухоли являются первым симптомом заболевания, что способствует раннему выявлению раков внутренних органов.
Дифференциальная диагностика проводится с ненаследственными опухолями кожи и внутренних органов.
При лечении опухолей кожи эффективны ароматические ретиноиды.
Профилактика заключается в медико-генетическом консультировании и обследовании членов семьи у онколога, а также ограничении воздействия на организм канцерогенных веществ.
Кожные заболевания, связанные с генетической нестабильностью
 Следует учитывать, что ряд наследственных синдромов, которые могли бы рассматриваться в этой группе болезней, в частности, атаксия-телеангиэктазия, относятся к группе первичных иммунодефицитных заболеваний.
Следует учитывать, что ряд наследственных синдромов, которые могли бы рассматриваться в этой группе болезней, в частности, атаксия-телеангиэктазия, относятся к группе первичных иммунодефицитных заболеваний.
Ниже приводятся особенности ряда генетических кожных заболеваний, связанных с генетической нестабильностью и ассоциирующихся со злокачественными новообразованиями кожи и внутренних органов.
Несмотря на существенный прогресс в понимании механизмов действия генов при целом ряде генетических заболеваний, связанных с высоким риском развития злокачественных новообразований, многие аспекты патогенеза рака при генитических синдромах все еще не ясны. В частности, трудно объяснить, почему при пигментной ксеродерме повышается частота только кожного рака, а при синдроме Блума, врождённом дискератозе, анемии Фанкони — в первую очередь злокачественных новообразований внутренних органов и лишь во вторую — рака кожи, тогда как синдром Коккейна и трихотиодистрофия, в некоторых случаях сопровождающиеся тем же дефектом репарации ДНК, как и пигментная ксеродерма, не сопровождаются повышением риска развития ни рака кожи, ни злокачественных новообразований внутренних органов.
Пигментная ксеродерма (ПК) — MIM 278700, 27872С 278730, 278740, 278760. Редкое наследственное аутосомно-рецессивное заболевание, соотношение полов 1:1. Отличается клинической и генетической гетерогенностью. Выделяют 7 комплементарных групп ПК: от А до G и так называемый вариант: XPV или пигментный ксеродермоид.
Генетические особенности пигментной ксеродермы:
|
Комплементарные группы |
Рак кожи |
Локус гена |
Особенность повреждения |
|
ПК-A OMIM 278700 |
++ |
9q34 |
Повреждение специфически ДНК-связанного протеина |
|
ПК-В OMIM 610651 |
+ |
2q21 |
ДНК хеликаза; ДНК-зависимая АТФаза |
|
ПК-С OMIM 278720 |
++ |
Зр25 |
ДНК эндонуклеаза |
|
ПК-D OMIM 178730 |
++ |
19.q13.2-q13.3 |
ДНК хеликаза; АТФ-аза транскрибирующий фактор |
|
ПК-Е OMIM 278740 |
+ |
11 р12—р11 |
ДНК-связывающий протеин |
|
ПК-F OMIM 278760 |
+ |
16р13.3-р13.13 |
Неизвестна |
|
ПК-G OMIM 278780 |
+ |
13q33 |
Однонитевая ДНК эндонуклеаза |
|
XPV OMIM 78750 |
+ |
6р21.1—р12 |
Доля ПК-A, ПК-С и XPV в общей структуре ПК составляет около 75%; ПК-D — 15%; ПК-В, ПК-Е, ПК-G и ПК-F — 10%.
Характеризуется повышенной фоточувствительностью, развитием пигментации и атрофии кожи, фотофобией, неврологической симптоматикой, прогрессирующим течением с очень высоким риском развития кожных опухолей. Повышенная чувствительность клеток к ультрафиолетовым лучам обусловлена нарушением репарации ДНК, возможна недостаточность эндонуклеазной эксцизии пиримидиновых димеров. Как и другие заболевания, связанные с нарушением репарации ДНК, ПК характеризуются нарушением активности естественных киллеров (NK) и продукции интерферона.
 Самый ранний клинический признак ПК — фотодерматит открытых участков кожи, возникающий даже при минимальной инсоляции и нередко сопровождающийся слезотечением и конъюнктивитом. Обычно эти изменения, соответствующие I клинической стадии ПК, возникают в период новорожденности или раннем детстве (до 2-3 лет) и в дальнейшем во II стадии прогрессируют с развитием сухости кожи, чешуек, гипо- и гиперпигментации по типу лентиго и веснушек, рубцевания, телеангиэктазий и атрофии, что создаёт картину пойкилодермии. Возможны также бородавчатые разрастания, трещины, изъязвления. Атрофия кожи лица сопровождается истончением кожи носа, ушных раковин, деформацией естественных отверстий, эктропионом, выпадением ресниц, изъязвлением слизистой оболочки. При ПК относительно часты неврологические нарушения, варьирующие от умеренных, определяемых только с помощью тестирования, до тяжёлых нарушений двигательных функций и задержки умственного развития или деменции.
Самый ранний клинический признак ПК — фотодерматит открытых участков кожи, возникающий даже при минимальной инсоляции и нередко сопровождающийся слезотечением и конъюнктивитом. Обычно эти изменения, соответствующие I клинической стадии ПК, возникают в период новорожденности или раннем детстве (до 2-3 лет) и в дальнейшем во II стадии прогрессируют с развитием сухости кожи, чешуек, гипо- и гиперпигментации по типу лентиго и веснушек, рубцевания, телеангиэктазий и атрофии, что создаёт картину пойкилодермии. Возможны также бородавчатые разрастания, трещины, изъязвления. Атрофия кожи лица сопровождается истончением кожи носа, ушных раковин, деформацией естественных отверстий, эктропионом, выпадением ресниц, изъязвлением слизистой оболочки. При ПК относительно часты неврологические нарушения, варьирующие от умеренных, определяемых только с помощью тестирования, до тяжёлых нарушений двигательных функций и задержки умственного развития или деменции.
Кожные раки возникают на III стадии ПК. Частота их развития в 5000 раз выше, чем в общей популяции. С первой опухолью обычно обращаются в детстве. При ПК развиваются все типы кожных раков: меланома (часто множественная), плоскоклеточный рак кожи, базалиома и др. Их анатомическая локализация подобна таковой в общей популяции, однако их течение неблагоприятно и отличается высокой смертностью от метастазов. Особенно высока предрасположенность к раку кожи при ПК-A, -С и -D; наоборот, при ПК-Е и -F он развивается редко и в более позднем возрасте; типы ПК-В и G редки и клинически проявляются синдромом Коккейна; тип XPV может сопровождаться как тяжёлыми, так и лёгкими поражениями кожи. При ПК развиваются и доброкачественные опухоли кожи: фибромы, кератоакантомы, ангиомы, кератомы, нейрофибромы, невромы, обызвествлённая эпителиома Малерба, атипичная фиброксантома и др. Злокачественный опухолевый процесс зависит от комплементарной группы: при ПК-Е варианте ПК преимущественно развивается базалиома, при ПК-С — плоскоклеточный рак кожи, при ПК-D — меланома. Интересно, что больные ПК имеют лишь незначительное повышение риска развития опухолей внутренних органов.
Гистологически в ранней стадии процесса изменения кожи аналогичны старческим (истончение эпидермиса, базофильная дистрофия коллагена, участки гиперкератоза) с высокой частотой признаков малигнизации; на опухолевой стадии гистологические изменения соответствуют типу новообразования.
Диагноз устанавливается на основании клинической картины и клинико-гениалогического анамнеза заболевания. Возможна ДНК-диагностика у плода.
 Дифференциальный диагноз проводится с наследственным лентиго, врожденным дискератозом, синдромом Ротмунда-Томсона.
Дифференциальный диагноз проводится с наследственным лентиго, врожденным дискератозом, синдромом Ротмунда-Томсона.
Тяжёлый прогноз обусловлен высокой склонностью к возникновению злокачественных новообразований кожи.
Лечение. Для профилактики развития рака кожи назначают интерфероны. Любые опухоли, в том числе и клинически доброкачественные, следует удалять на самых начальных этапах развития. Помимо хирургического удаления применяют криодеструкцию, удаление опухоли лучами лазера, назначают внутрь ароматические ретиноиды, аппликации мазей с цитостатиками (5-фторурацилом и др.) или 5% крема имиквимод.
Профилактика заключается в недопущении кровных браков. Для предотвращения развития кожных раков важно раннее выявление заболевания и защита от воздействия на кожу ультрафиолетовых лучей (солнцезащитные кремы и др.); рекомендуется также длительный прием ретиноидов.
Альбинизм кожноглазной MIM 203200, 203100, 203290. Альбинизм кожноглазной (АК) — это гетерогенное заболевание, характеризующееся отсутствием или резким уменьшением пигмента в коже, волосах, радужной оболочке глаз. Выделяют две формы ОК — тирозиназоотрицательную и тирозиназоположительную, патогенез которых связан с отсутствием или недостаточной активностью тирозиназы. Высокая частота кожных раков при АК обусловлена повреждением ДНК ультрафиолетовыми лучами в связи с отсутствием меланина.
Клиническая картина характеризуется тотальной депигментацией кожи, волос, оболочек глаз, которая выявляется сразу после рождения ребёнка, так как именно с этого времени повышена чувствительность кожи к солнечным лучам (солнечные ожоги даже при незначительной инсоляции, солнечный хейлит, фотофобия). Кожа обычно имеет молочно-белый цвет, суховата, потоотделение снижено, имеется гипотрихоз. Отмечается снижение резистентности к инфекциям, глухота, олигофрения, эпилепсия, бесплодие. У больных, не достигших 20-летнего возраста, нередки актинический кератоз; позже развиваются очаги атрофии кожи, сенильный эластоз, телеангиэктазии. При кожноглазном альбинизме также повышена частота немеланотических раков кожи, чаще плоскоклеточного рака (нередко достигающего диаметра более 4 см), чем базалиомы. Нередко плоскоклеточный рак кожи метастазирует и сопровождается летальным исходом. Повышена также частота беспигментных меланоцитарных невусов и амеланотической меланомы.
Диагноз устанавливается на основании клинической картины заболевания и ДНК-диагностики.
Лечение АК неэффективно. Назначают фотозащитные кремы. Для профилактики плоскоклеточного рака кожи и базалиомы следует избегать длительного пребывания на солнце, пользоваться широкополыми шляпами, наружно применять ароматические ретиноиды. Больные должны ежегодно осматриваться дерматоонкологом для раннего выявления злокачественных опухолей кожи.
 Эпидермодисплазия верруциформная Левандовского-Лютца (ЭВЛЛ) (син.: verrucosis generalisata) — MIM 305350, 605828, 605829, 226400
Эпидермодисплазия верруциформная Левандовского-Лютца (ЭВЛЛ) (син.: verrucosis generalisata) — MIM 305350, 605828, 605829, 226400
Редкое, обычно аутосомно-рецессивно наследуемое заболевание с уникальной предрасположенностью к инфицированию кожи специфическими разновидностями ВПЧ. Проявляется в раннем детстве. У больных ЭВЛЛ выделены несколько типов ВПЧ, которые можно разделить на две группы: с высоким онкогенным потенциалом — 5, 8 и 47-й типы, которые обнаруживаются в 90% плоскоклеточных раков кожи, развившихся на фоне очагов ВЭ; с низким злокачественным потенциалом — 3, 14, 20, 21 и 25-й типы, которые обычно обнаруживаются в доброкачественных очагах ЭВЛЛ.
Генетические дефекты и наличие ВПЧ (чаще 5-го типа) у больных ЭВЛЛ обычно ассоциируются с нарушением клеточного звена иммунитета (подавление иммунного ответа и нарушение клеточноопосредованной цитотоксичности, хотя антигенпрезентирующая активность клеток Лангерганса не нарушена) и воздействием таких канцерогенных факторов, как ультрафиолетовое излучение спектра В и ионизирующая радиация.
Клинически характеризуется множеством пятен и бородавок, главным образом плоского типа, которые имеют тенденцию к слиянию и распространяются иногда на всю поверхность предплечий, голеней, лица. В отдельных случаях формируются участки ограниченного гиперкератоза, дисхромии, алопеции. У 30-50% больных ЭВЛЛ, особенно на открытых участках кожи развиваются болезнь Боуэна, бовеноидный папулёз с трансформацией в плоскоклеточный рак, в 20-30% случаев, или базалиома. Первый очаг плоскоклеточного рака кожи возникает в среднем до 30 лет. Развитие его сопровождается усилением роста бородавчато-подобных элементов, слиянием их в крупные инфильтрированные бляшки, которые приобретают красный или красно-коричневый цвет, изъязвлением. Плоскоклеточные раки, развивающиеся при ЭВЛЛ, обычно характеризуются периневральной инвазией и частым метастазированием в лимфатические узлы, а иногда и во внутренние органы, что приводит к летальному исходу. Средний возраст метастазирования — 34 года. Следует учитывать, что агрессивному клиническому течению рака кожи у больных ЭВЛЛ способствует рентгенотерапия. В ряде случаях отмечается длительная трансформация одного или нескольких элементов в базалиому.
Лечение. Бородавчатые элементы (в том числе предраковые) удаляют методами криодеструкции, электрокоагуляции, с помощью лучей ниодимового лазера, фотодинамической терапии; наружных препаратов ретинойной кислоты (0,05-0,1%), 5% крема с 5-фторурацилом или 5% крема имиквимод. При раке кожи на фоне ЭВЛЛ хирургическое удаление опухоли проводится в комбинации с химиотерапией (проспидин, блеомицин). Эффективны ароматические ретиноиды (ацитретин) в комбинации с интерфероном.
Профилактика злокачественной трансформации ЭВЛЛ заключается в использовании солнцезащитных средств. Учитывая важную роль в развитии озлокачествления ВПЧ-5, целесообразно повышение иммунологической реактивности организма как больных, так и их ближайших родственников с использованием препаратов интерферона. Целесообразно медико-генетическое консультирование больных и их ближайших родственников перед вступлением в брак.
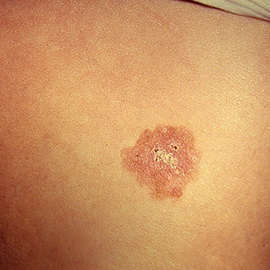
Множественная самоизлечивающаяся плоскоклеточная эпителиома Фергюссона-Смита — MIM 132800. Редкое наследственное заболевание; тип наследования аутосомно-доминантный. Предполагается, что ген, определяющий развитие заболевания, картируется в хромосомном локусе 9q31.
При этом заболевании происходит быстрое развитие кератоакантома -подобных узлов на лице и других облучаемых солнцем участках кожи. Хотя возраст больных варьирует от 8 до 62 лет, чаще заболевание встречается в раннем подростковом или юношеском возрасте.
Кожные опухоли представлены множественными куполообразными узлами с центральным кератиновым кратером, которые в течение нескольких недель увеличиваются, изъязвляются и подвергаются спонтанной инволюции с образование вдавленных рубчиков неправильной формы. Если клинически элементы напоминают кератоакантому, то по гистологическому строению они нередко неотличимы от плоскоклеточного рака кожи. Изредка они метастазируют.
 Синдром Ротмунда-Томсона (СРТ) (син.: врождённая пойкилодермия) — MIM 268400. Редкий наследственный синдром, тип передачи аутосомно-рецессивный. Частота поражения лиц обоего пола примерно одинаковая. В некоторых случаях выявлены мутации в гене RECQL4. Отмечено снижение репарации ДНК в культивированных фибробластах после УФ-С и гамма-облучения. Наличие хромосомной нестабильности предполагается исходя из высокой частоты ассоциации синдрома со злокачественными новообразованиями.
Синдром Ротмунда-Томсона (СРТ) (син.: врождённая пойкилодермия) — MIM 268400. Редкий наследственный синдром, тип передачи аутосомно-рецессивный. Частота поражения лиц обоего пола примерно одинаковая. В некоторых случаях выявлены мутации в гене RECQL4. Отмечено снижение репарации ДНК в культивированных фибробластах после УФ-С и гамма-облучения. Наличие хромосомной нестабильности предполагается исходя из высокой частоты ассоциации синдрома со злокачественными новообразованиями.
Кожные поражения возникают в первые 3-6 мес. и проявляются диффузной эритемой, иногда отёком и везикуляцией открытых участков кожи: лица, ушных раковин, тыла кистей, разгибательной поверхности конечностей. Позже на этих же местах возникают явления пойкилодермии (телеангиэктазии, точечные участки гипо- и гиперпиг ментации, атрофии), напоминающие проявления хронического радиационного дерматита. В дальнейшем сыпь может распространяться на ягодицы, шею, туловище. Прогрессирование этих поражений продолжается до возраста 3-5 лет. На втором десятилетии жизни у больных развиваются кератотические и бородавчатые высыпания на кистях, предплечьях, стопах и голенях. С возрастом фоточувствительность уменьшается, но сформировавшиеся в детстве высыпания персистируют в течение всей жизни. Для СРТ также характерны: ювенильная катаракта, дистрофия волос (разрежение или отсутствие волос на голове, бровях, ресницах, лобке, в подмышечных впадинах) и ногтей, гипоплазия зубов с высокой частотой кариеса, нарушение физического развтия, костные деформации, эндокринные расстройства, гипогонадизм в сочетании с низким ростом, нарушение потоотделения и т.д.
СРТ ассоциируется с развитием злокачественных новообразований внутренних органов (остеосаркома, рак желудка, лейкоз и т.д.) и немеланотических раков кожи: множественных очагов болезни Боуэна, плоскоклеточного рака кожи, базалиомы, возникающих на фоне очагов гиперкератоза и атрофии.
Гистологически у детей при СРТ обнаруживают уплощение и атрофию эпидермиса с отёком дермо-эпидермального соединения, иногда — некоторое расширение сосудов дермы и их периваскулярную инфильтрацию лимфоцитами; на открытых участках кожи у взрослых — комбинацию фрагментации эластических волокон дермы с явлениями пятнистого бовеноидного дискератоза в эпидермисе.
Дифференциальный диагноз проводится с синдромом Коккейна, синдромом Вернера, врождённым дискератозом, прогерией, гипогидротической эктодермальной дисплазией, синдромом Блума.
Лечение гиперкератотических, бородавчатых высыпаний и рака кожи проводится хирургически, с помощью криодеструкции, лучей лазера или мазей с цитостатическими препаратами (50% проспидиновой, 5% флюороурациловой), приёмом внутрь ароматических ретиноидов. При лечении телеангиэктазий в области лица используют аргоновый лазер, а также ароматические ретиноиды.
В целях профилактики рака кожи важное значение имеет ее защита от солнечного облучения. Лица пожилого возраста должны консультироваться дерматоонкологом с целью раннего выявления злокачественных опухолей кожи.
 Дискератоз врождённый (ДВ) — MIM 305000, 242230, 127550. Редкая форма эпидермальной дисплазии. Встречается почти исключительно у мужчин. Отмечено три типа наследования: аутосомно-рецессивный, Х-сцепленный с полом рецессивный и аутосомно-доминантный. Ген ДВ располагается на длинном плече Xq28. У больных ДВ выявляются различные иммунные нарушения.
Дискератоз врождённый (ДВ) — MIM 305000, 242230, 127550. Редкая форма эпидермальной дисплазии. Встречается почти исключительно у мужчин. Отмечено три типа наследования: аутосомно-рецессивный, Х-сцепленный с полом рецессивный и аутосомно-доминантный. Ген ДВ располагается на длинном плече Xq28. У больных ДВ выявляются различные иммунные нарушения.
Дети с этим синдромом остаются здоровыми до 5-летнего возраста. В последующие 10 лет у них развиваются эритема, сетчатая гиперпигментация и телеанги-эктазии, наиболее выраженные на лице, шее и бёдрах, а также ониходистрофия, оральная или аногенитальная лейкоплакия. У детей чаще имеются телеангиэктазии и атрофия кожи с исчезновением ладонной дерматоглифики, дистрофия ногтей и паронихии, в возрасте 3-5 лет возникают изменения пигментации в виде сетчатой серо-коричневой пигментации, особенно на шее, бёдрах и груди, наличие атрофий и телеангиэктазий в этих местах придают ей картину пойкилодермии; у пожилых — ладонно-подошвенный гиперкератоз, гипергидроз, геморрагические пузыри на слизистой оболочке рта и пойкилодермически изменённой коже. Также нередки нарушения со стороны глаз (блефарит, эктропион, катаракта, глаукома, обструкция выходных отверстий слёзных протоков), гингивиты, лейкокератоз щёк, языка, нёба, раннее выпадение зубов, реже — маленький рост и костные нарушения, ранняя алопеция, преждевременная седина; у 30% -умственная отсталость. Системные проявления: гематологические заболевания (спленомегалия, тромбоцитопения, апластическая анемия и панмиелофтиз). Лейкоплакия у больных ВД нередко трансформируется в плоскоклеточный рак. Заболевание также ассоциируется со злокачественными опухолями ротоглотки, аногенитальной области и желудочно-кишечного тракта, описано развитие лимфогранулематоза, аденокарциномы поджелудочной железы.
При гистологическом исследовании выявляют атрофию эпидермиса, увеличение количества меланина в базальном слое; в дерме — расширение сосудов и появление меланофор.
Дифференциальный диагноз проводится с синдромом Ротмунда-Томсона, пигментной ксеродермой, ангидротической эктодермальной дисплазией.
Прогноз — неблагоприятный из-за развития гематологических заболеваний и рака на фоне лейкоплакии. Причиной летального исхода могут быть инфекции и желудочно-кишечные кровоизлияния.
Лечение опухолей кожи проводят хирургически или аппликациями мазей с цитостатиками (5% 5-фторурациловая, 50% проспидиновая и др.); лечение лейкоплакии проводят этретинатом, что предотвращает её частую в трансформацию плоскоклеточный рак.
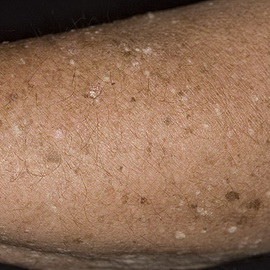
 Анемия Фанкони (АФ) — MIM 227650. Редкое наследственное заболевание, передающееся по аутосомно-рецессивному типу. Мутация в любой группе генов FANC А, В, С, Dl, D2, Е, F, G, I, L, М, N приводит к АФ. Различают четыре группы, причем ген группы С клонирован и картирован на хромосоме 9q22.3, а ген группы А находится на 16q24.3. Хотя функция белка, закодированного в этом локусе, неизвестна, предполагается, что он является цитоплазматическим и играет прямую роль в репарации ДЕ1К. Клетки АФ приводят к хромосомной нестабильности, сопровождающейся повышеной тенденцией к разрыву ДЕ1К перекрёстносвязывающими агентами, такими как диэпоксибутан, используемый в диагностическом тесте на АФ.
Анемия Фанкони (АФ) — MIM 227650. Редкое наследственное заболевание, передающееся по аутосомно-рецессивному типу. Мутация в любой группе генов FANC А, В, С, Dl, D2, Е, F, G, I, L, М, N приводит к АФ. Различают четыре группы, причем ген группы С клонирован и картирован на хромосоме 9q22.3, а ген группы А находится на 16q24.3. Хотя функция белка, закодированного в этом локусе, неизвестна, предполагается, что он является цитоплазматическим и играет прямую роль в репарации ДЕ1К. Клетки АФ приводят к хромосомной нестабильности, сопровождающейся повышеной тенденцией к разрыву ДЕ1К перекрёстносвязывающими агентами, такими как диэпоксибутан, используемый в диагностическом тесте на АФ.
АФ встречается у детей в возрасте от 4 до 10 лет. Характеризуются аплазией костного мозга и панцитопенией (анемия, нейтроцитопения, тромбоцитопения) в сочетании с рядом соматических и метаболических нарушений: задержкой роста, дефектами формирования скелета (микроцефалией и др.), глаз, ушных раковин, сердца, нервов, мочеполового и желудочно-кишечного трактов, гипогонадизмом, гипоплазией почек или селезёнки, аминоацидурией, глюкозурией.
Кожный процесс представлен оливково-коричневыми пятнами на нижней части туловища, сгибах конечностей, шее. В пределах этих очагов разбросаны мелкие депигментированные и гиперпигментированные пятна, придающие ему сетчатый вид. Кроме того, имеются пятна типа «кофе с молоком».
У больных АФ отмечается заметная предрасположенность к злокачественным новообразованиям, особенно к острому миелолейкозу, который в 52% случаях развивается в возрасте до 40 лет. Также часты плоскоклеточный рак кожи, раки ротоглотки, пищеварительного, мочеполового трактов, которые могут развиваться в раннем возрасте.
Синдром Шпиглера-Брока (СШБ) — MIM 605041. Характеризуется сочетанием трихоэпителиом, цилиндром и спираденом. Опухоли обычно множественные и возникают в пубертатном периоде, чаще у женщин. Семейные случаи множественной трихоэпителиомы связывают с аутосомно-доминантным типом наследования. При этом члены одной семьи имеют различные проявления синдрома. Солитарные очаги обусловлены спорадическими мутациями в гене 16ql2-ql3.
Клинически синдром характеризуется множественными трихоэпителиомами, представленными мелкими (около 0,5 см в диаметре) прозрачными (напоминающими жемчужины) куполообразными папулами, расположенными в центре лица, преимущественно в носогубных складках и периорбитально (изредка на волосистой части головы и туловище).
С возрастом количество опухолей увеличивается, они могут достигать больших размеров и изъязвляются, приобретая сходство с базалиомой.
При гистологическом исследовании выявляются кистозные полости, заполненные роговыми массами, по периферии которых отмечается пролиферация базалоидных клеток. Положительная реакция на щелочную фосфотазу помогает выявлению в опухоли рудиментарных волосяных сосочков.
Диагноз устанавливается по данным клиники и результатам гистологического исследования.
Дифференциальный диагноз проводится с базалиомой, аденомой сальных желез, злокачественной цилиндромой, сирингомой.
Лечение заключается в удалении опухоли (хирургическом, с использованием дермабразии или лазера).
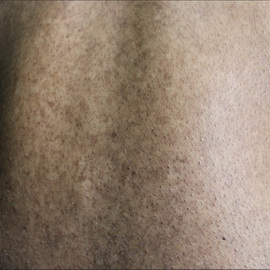 Хабера синдром (XC). Очень редкое наследственное заболевание, проявляющееся стойкой розацеаподобной сыпью, интраэпидермальными опухолями кожи и повышенной чувствительностью к солнцу. Тип наследования аутосомно-доминантный. Генетическая основа неизвестна. Впервые описан у пяти членов одной семьи.
Хабера синдром (XC). Очень редкое наследственное заболевание, проявляющееся стойкой розацеаподобной сыпью, интраэпидермальными опухолями кожи и повышенной чувствительностью к солнцу. Тип наследования аутосомно-доминантный. Генетическая основа неизвестна. Впервые описан у пяти членов одной семьи.
Начинается в раннем детском возрасте, когда под влиянием солнечных лучей возникает стойкая эритема и зуд в области щёк, носа и подбородка. На этом фоне возникают телеангиэктазий и мелкие плотные фолликулярные папулы красного цвета, участки шелушения, мелкие рубчики. На закрытых участках кожи (грудь, спина, волосистая часть головы на границе с гладкой кожей) формируются симметричные гиперкератотические и бородавчатые очаги диаметром до 1 см с выраженной тенденцией к трансформации в болезнь Боуэна.
При гистологическом исследовании этого генетического заболевания кожи выявляют акантоз, паракератоз, участки спонгиоза, увеличение митотической активности клеток базального слоя, стойкое расширение и увеличение количества сосудов дермы, пролиферацию недифференцированных клеток сальных желёз, участки фиброза. В очагах поражения на туловище обнаруживают веррукозный гиперкератоз, диспластический кератоз и внутриэпидермальный рак; инвазивный рак не описывается.
Лечение бородавчатых элементов проводят с помощью рентгенотерапии; новообразования кожи удаляют хирургически.
KID-синдром (син.: kereatitisichtiosisdeafness) — MIM 148210. Различно наследуемый ихтиозиформный синдром, сопровождающийся генерализованным ихтиозом, кератитом и нейросенсорной глухотой, иногда с нарушением физического и умственного развития. Кожные изменения включают также веррукозные бляшки на подбородке, носу, ушных раковинах, ладонно-подошвенную кератодермию, очаговую или диффузную алопецию, дистрофию ногтей, поражение волос и потовых желёз, частые бактериальные и грибковые инфекции. В раннем детстве может встречаться плоскоклеточный рак кожи, который развивается, как правило, на фоне хронически инфицированных незаживающих ран. В таких случаях его следует гистологически дифференцировать с псевдокарциноматозной гиперплазией эпидермиса.
Больные KID-синдромом нуждаются в периодических осмотрах дерматоонколога с повторными биопсиями длительно незаживающих язв.
Хронический (гранулематозный) кожно-слизистый кандидоз (ХГКСК) (син.: хронический генерализованный кандидоз, кандидозная гранулёма, эндокринно-кандидозный синдром) -MIM 212050, 114580, 240300
Формируется у лиц с врождённых иммунодефицитом. Выделяют 4 клинические группы ХГКСК:
- Семейный аутосомно-рецессивный (MIM 21205) и аутосомно-доминантный (MIM 114580) ХКСК, включающий мягкий оральный кандидоз и несколько кожных очагов.
- ХКСК с ранним началом, диффузным тяжёлым поражением кожи и слизистых оболочек с формированием гранулём.
- Семейный ХКСК (MIM 240300), сопровождающийся эндокринопатией (чаще всего гипопаратиреозом, сахарным диабетом, заболеваниями щитовидной железы и надпочечников).
- ХКСК с поздним началом, сопровождающийся злокачественными новообразованиями, чаще — тимомой; именно у больных этой группы, как правило, в молодом возрасте, может развиться плоскоклеточный рак слизистой оболочки рта, и нередко он метастазирует и сопровождается летальным исходом; развитие его связывают с частым рецидивирующим поражением слизистых оболочек, замедленным заживлением обусловленных им эрозий и язв, а также иммунными нарушениям.
Диагноз устанавливается на основании клинической картины и лабораторного выявления дрожжеподобных грибов рода Candida.
Лечение проводится системными антимикотическими препаратами (орунгал и т. д.)
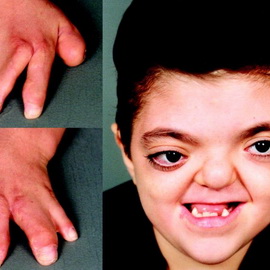
 Синдром Горлина-Гольтца (синдром невоидной базально-клеточной карциномы; невобазалиоматоз, синдром базальноклеточных невусов; синдром Горлина) — MIM 109400. Генетически детерминированный полиорганный синдром, наследуемый по аутосомнодоминантному типу с высокой пенетрантностью и различной экспрессивностью.
Синдром Горлина-Гольтца (синдром невоидной базально-клеточной карциномы; невобазалиоматоз, синдром базальноклеточных невусов; синдром Горлина) — MIM 109400. Генетически детерминированный полиорганный синдром, наследуемый по аутосомнодоминантному типу с высокой пенетрантностью и различной экспрессивностью.
Характеризуется сочетанием базалиом с разнообразными пороками развития скелета, глаз, нервной системы и с опухолями различной локализации. Причём ассоциация синдрома Горлина-Гольтца с различными опухолями обусловлена мутациями в генах РТСН1, РТСН2 или SUFU. Почти все изменения — врождённые. Нами выявлена связь синдрома Горлина-Гольтца с антигенами HLA-A10 и В14.
Базалиомы также могут быть врождёнными, хотя чаще появляются в позднем детском возрасте, но обычно до 35 лет. Как правило, они множественные, располагающиеся симметрично и билатерально на открытых и на закрытых участках кожи и преимущественно поражают лицо, шею, туловище, конечности. Количество их может достигать нескольких сотен. Вначале это поверхностные базалиомы диаметром 1-3 см, развивающиеся медленно и практически не изменяющиеся в размере вплоть до 2-3-го десятилетия жизни. После чего некоторые из них быстро увеличиваются до 5-10 см в диаметре и под влиянием неблагоприятных внешних факторов среди них появляются кистозные, язвенные формы, а также метатипический рак кожи.
Течение базалиом при синдроме Горлина-Гольтца, в отличие от первичномножественных базалиом, более агрессивное и резистентное к лечению, они чаще рецидивируют и могут метастазировать. Изредка опухоли развиваются из предшествующих ладонно-подошвенных углублений.
Отсутствие базалиом и других опухолей кожи при синдроме Горлина-Гольтца бывает редко. Точечные пигментированные углубления на ладонях и подошвах нередко появляются раньше других кожных проявлений синдрома и ассоциируются с ним в 70-80% случаев, на дне углублений почти всегда видны телеангиэктазий. Количество углублений (диагностическое количество равно 3) иногда достигает нескольких сотен; больше всего их на боковых поверхностях ладоней, подошв и пальцев кистей. Развитие из связывают с преждевременным слущиванием рогового слоя эпидермиса. Синдром Горлина-Гольтца также может ассоциироваться с такими поражениями кожи, как эпидермальные кисты, милиум, фиброма, липома, пигментные невусы, веснушки, ладонно-подошвенный гиперкератоз, комедоны.
Среди костных аномалий, которые имеют врождённый характер и выявляются при этом синдроме в 75-90% случаев, наиболее часты множественные одонтогенные кисты верхней и нижней челюстей. Их нагноение (сопровождающееся отеком лица и тупой болью) заканчивается вскрытием в полость рта. Кроме того, описываются такие костные аномалии, как кифоз, сколиоз, расщепление рёбер, воронкообразная грудная клетка, укорочение IV пястных костей, синостозы, прогнатия, парадонтоз, неправильное расположение зубов, готическое нёбо, срединный носовой синус, широкие носовые ходы, истинный гипертелоризм, выступающие лобные бугры, дизостозы костей лицевого черепа, субкорнеальные кистозные изменения длинных трубчатых и плоских костей.
Аномалии глаз встречаются в 26% случаев и проявляются врождённой слепотой, дистопией внутренних углов глаза, катарактой, глаукомой, колобомой, страбизмом.
Поражение ЦНС отмечаются у 10-42% больных и проявляются гидроцефалией, микроцефалией, недоразвитием мозолистого тела головного мозга, пластинчатого обызвествления серпа большого мозга, кальцификацией твёрдой и мягкой мозговых оболочек, эпилепсией, деменцией, задержкой психического развития.
Примерно в 17% случаев имеет место патология эндокринной (акромегалия, тиреоидит) и половой (крипторхизм, гипогонадизм, бесплодие, двурогая матка, фиброматоз яичников) систем.
Синдрому могут сопутствовать и другие аномалии развития, такие как врождённое отсутствие почки и мочеточника или анемия Фанкони.
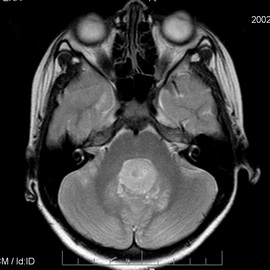 Синдром Горлина — Гольтца ассоциируется с такими опухолями, как медулло-эластома, фиброматоз яичников, фибросаркома челюстей, фиброма, тератома и цистаденома яичников, лейомиома, нейрофиброматоз I типа, лимфома Ходжкина, ретинобластома, менингиома, рабдомиома, множественные миомы матки, сеиинома, рак матки, ренин-секретирующая опухоль яичника, опухоль мозжечка, ненингиома. В частности, медуллобластома чаще поражает мальчиков в возрасте до 2 лет и, таким образом, нередко возникает до появления кожных опухолей.
Синдром Горлина — Гольтца ассоциируется с такими опухолями, как медулло-эластома, фиброматоз яичников, фибросаркома челюстей, фиброма, тератома и цистаденома яичников, лейомиома, нейрофиброматоз I типа, лимфома Ходжкина, ретинобластома, менингиома, рабдомиома, множественные миомы матки, сеиинома, рак матки, ренин-секретирующая опухоль яичника, опухоль мозжечка, ненингиома. В частности, медуллобластома чаще поражает мальчиков в возрасте до 2 лет и, таким образом, нередко возникает до появления кожных опухолей.
Диагноз устанавливается на основании совокупности клинико-гистологических данных и результатов обследования стоматолога, челюстно-лицевого хирурга, невропатолога, окулиста, эндокринолога, гинеколога, рентгенографии плоских костей, а для выявления пластинчатого обызвествления серпа большого мозга — рентгенологического исследования черепа.
Дифференциальный диагноз этой генетической болезни кожи проводят с первичными множественными базалиомами.
Течение сопровождается появлением новых опухолей в течение всей жизни. Прогноз лучше, чем в случаях с медуллобластомой, возникающей независимо от синдрома Горлина-Гольтца.
Лечение. Опухоли, расположенные в центральной части лица, наушных раковинах или в их окружности, подлежат иссечению по методу Mohs — с микроскопией замороженных горизонтальных срезов для определения объёма операции. Мелкие опухоли на туловище и конечностях удаляют методом электрокоагуляции, кюретажа, фотодинамической терапии (с наружным нанесением фотосенсибилизатора), используют также внутриопухолевое введение интерферона-а2b, комбинацию ароматических ретиноидов, аппликации фторурацила (5% крем в течение 25-30 суток) или 5% крема имиквимод.
В профилактических целях эффективен прием внутрь ароматических ретиноидов.
Источник статьи
Предыдущее: Как отходят воды у беременных перед родами